


Seong-Jae Yoo, PhD, & Gabriel Giancaspro, PhD, U.S. Pharmacopeial Convention (USP) 03.01.17
Almost a decade has passed since the FDA current Good Manufacturing Practice regulations (cGMPs) for dietary supplements (21 CFR Part 111) were promulgated. Unfortunately, as recent results of FDA GMP inspections indicate, many dietary supplement manufacturers are not fully complying with cGMPs, in many cases due to lack of specifications for ingredients and finished products. Monographs of the United States Pharmacopeia (USP) can be useful and important resources for those having to comply with cGMPs.
GMP Background
cGMPs were introduced as a regulatory tool with the intent to ensure the safety and quality of dietary supplement products through manufacturing rules. Products manufactured in violation of cGMPs are considered adulterated as a matter of law regardless of whether the product has any defects.
The key principle of GMP compliance is that finished product quality needs to be built into the manufacturing process. Each step of the manufacturing process needs to be controlled to lower the risks that the finished product will not comply with the established quality specifications. Congress has given FDA the authority to regulate the manufacturing processes, inspect manufacturing facilities and pursue enforcement actions against regulated firms, individuals and products that fail to comply with cGMPs. Among other measures, FDA compliance and enforcement actions can include seizures, injunctions, import alerts and criminal investigation.
Observations & Failures
cGMPs make the dietary supplement manufacturers responsible for determining the appropriate quality parameters for their finished products, and mandate the establishment of specifications for identity, purity, strength and composition, as well as limits on potential contaminants. However, since the inception of the cGMPs, FDA inspectors have observed more than 9,000 cGMP compliance violations in about 2,000 inspection citations. Approximately 27% of the observations involved the failure to establish an appropriate production and process control system (as specified in Subpart E of cGMPs), including the failure to establish appropriate specifications (§ 111.70), along with a failure to comply with component identity verification requirements (§ 111.75).
Observations regarding the failure to establish specifications (§ 111.70) accounted for about 12% of the total observations cited in 2015, according to FDA inspection data. Observations related to component specifications typically stated “…You failed to establish specifications for identity, purity, strength, composition, and limits on contaminants for each component used in the manufacture of your dietary supplements, as required by 21 CFR 111.70(b)….”; those related to finished dietary supplements typically indicated “…You failed to establish product specifications for the identity, purity, strength, and composition of the finished batch of the dietary supplement, and for limits on those types of contamination that may adulterate, or that may lead to adulteration of, the finished batch of the dietary supplement, as required by 21 CFR 111.70(e)....” As repeatedly described in warning letters, properly addressing the five test specifications of “identity, purity, strength, composition, and limits on contaminants” is imperative for both components and finished products.
Defining Quality
In the Preamble to the final rule for 21 CFR Part 111, FDA defines quality for dietary supplements: ‘‘Quality means that the dietary supplement consistently meets the established specifications for identity, purity, strength, and composition and has been manufactured, packaged, labeled, and held under conditions to prevent adulteration under section 402(a)(1), (a)(2), (a)(3), and (a)(4) of the [Federal Food, Drug, and Cosmetic Act].’’
Although warning letters from FDA inspectors repeatedly place emphasis on the five attributes for which specifications must be set (i.e., identity, purity, strength, composition and limits on contaminants), FDA does not provide a specific definition for the meaning of the terms “identity,” “purity,” “strength,” and “composition” in the final rule. According to the Preamble, these terms were not defined because “[t]he exact way in which the dietary supplement industry uses these terms may vary, and defining these terms could limit the flexibility that is needed to accommodate such variations.” Instead of defining the terms, FDA provided interpretations and examples that should be considered when establishing specifications. Table 1 lists the explanatory text from the Preamble (without the specific examples that were provided).
Despite the agency’s intent to allow some flexibility in the use of these terms, the interpretations provided in the Preamble may not necessarily provide the level of clarity desired by the industry. The agency’s interpretations of these terms were set in the context of finished dietary supplements, and may be extended to components, insofar as the specifications of components affect the specifications of the final dietary supplements. USP suggests that further explanation of these terms would be useful, particularly in the context of defining quality attributes for the different classes of components found in dietary supplements. Some other sections of the Preamble to the cGMP final rule clarify the use of these terms as applied to the ingredients (see Table 2).
Terms for Quality Attributes
USP sets standards for the identity, strength, quality, and purity of medicines, food ingredients, and dietary supplements manufactured, distributed and consumed worldwide. USP standards for drugs and dietary supplements are recognized in U.S. federal law and are enforceable by FDA. However, although it is mandatory for drug product manufacturers to comply with USP standards, dietary supplement standards developed by USP are voluntary.
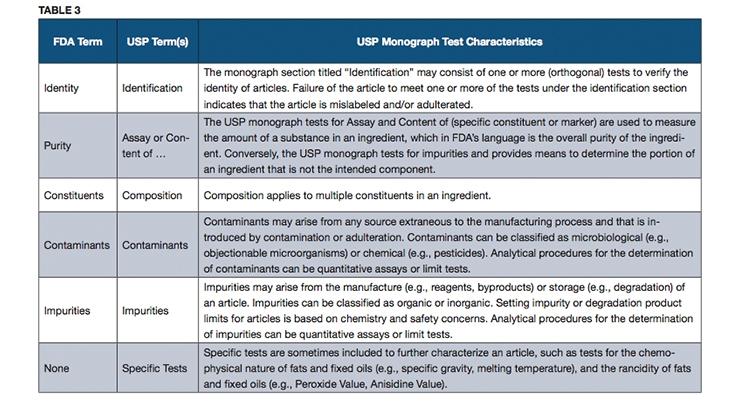
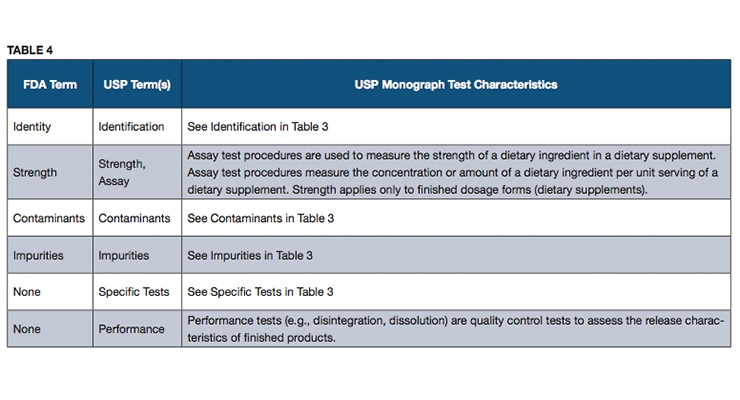
An official USP quality standard for an article (i.e., excipient, dietary ingredient or dietary supplement), referred to as a monograph, sets forth the article’s name, definition, specifications (i.e., tests, test procedures and acceptance criteria) and other requirements related to packaging, storage and labeling. USP monographs include specifications for identity, assay, strength, composition, limits for contaminants, specific tests and/or performance criteria (primarily for finished dosage units). Some of the terms used in USP monographs may differ from the FDA terms, and yet the specifications that they represent are intended to address all of the quality attributes mentioned in FDA’s cGMP requirements, as described in Tables 3 and 4.
In general, USP monographs do not refer to “purity” as a test in a monograph, but rather refer to an “Assay” or “Content” test procedure that can measure the overall purity as referred to in FDA’s cGMPs when applied to an ingredient. The test for strength in a USP monograph is used to measure the amount of a dietary ingredient per unit of measure in a dietary supplement, and it has the same meaning as in FDA’s cGMPs. The term “Composition” in a USP monograph is used to refer to those tests for multiple constituents in a complex dietary ingredient.
USP monographs may include specific tests for an article, such as loss on drying, or residue on ignition. Performance tests such as disintegration or dissolution are performed on a dietary supplement to help ensure that the supplement will release the ingredients properly to make them available for absorption by the body or to exert their function as claimed.
The type of performance test used may be dependent on health considerations (e.g., dissolution test for folic acid in supplements). In conclusion, USP monographs can serve as useful resources to help dietary supplement manufacturers comply with cGMPs when setting quality specifications. Manufacturers can benchmark their tests, analytical procedures, and acceptance criteria against those in USP monographs when setting a specification for a component (e.g., excipient, vitamin, mineral, amino acid, specialty chemical, botanical) or a finished product (e.g., tablet, capsule, or soft gel). USP official standards for dietary supplements offer manufacturers a scientifically valid means of supporting compliance with the specification requirements described in 21 CFR 111.70. Knowledge of FDA’s cGMP requirements related to specifications for dietary supplements—coupled with a robust understanding of how USP monographs support the regulatory regime—can help enhance the quality of the dietary supplement marketplace and protect public health.
GMP Background
cGMPs were introduced as a regulatory tool with the intent to ensure the safety and quality of dietary supplement products through manufacturing rules. Products manufactured in violation of cGMPs are considered adulterated as a matter of law regardless of whether the product has any defects.
The key principle of GMP compliance is that finished product quality needs to be built into the manufacturing process. Each step of the manufacturing process needs to be controlled to lower the risks that the finished product will not comply with the established quality specifications. Congress has given FDA the authority to regulate the manufacturing processes, inspect manufacturing facilities and pursue enforcement actions against regulated firms, individuals and products that fail to comply with cGMPs. Among other measures, FDA compliance and enforcement actions can include seizures, injunctions, import alerts and criminal investigation.
Observations & Failures
cGMPs make the dietary supplement manufacturers responsible for determining the appropriate quality parameters for their finished products, and mandate the establishment of specifications for identity, purity, strength and composition, as well as limits on potential contaminants. However, since the inception of the cGMPs, FDA inspectors have observed more than 9,000 cGMP compliance violations in about 2,000 inspection citations. Approximately 27% of the observations involved the failure to establish an appropriate production and process control system (as specified in Subpart E of cGMPs), including the failure to establish appropriate specifications (§ 111.70), along with a failure to comply with component identity verification requirements (§ 111.75).
Observations regarding the failure to establish specifications (§ 111.70) accounted for about 12% of the total observations cited in 2015, according to FDA inspection data. Observations related to component specifications typically stated “…You failed to establish specifications for identity, purity, strength, composition, and limits on contaminants for each component used in the manufacture of your dietary supplements, as required by 21 CFR 111.70(b)….”; those related to finished dietary supplements typically indicated “…You failed to establish product specifications for the identity, purity, strength, and composition of the finished batch of the dietary supplement, and for limits on those types of contamination that may adulterate, or that may lead to adulteration of, the finished batch of the dietary supplement, as required by 21 CFR 111.70(e)....” As repeatedly described in warning letters, properly addressing the five test specifications of “identity, purity, strength, composition, and limits on contaminants” is imperative for both components and finished products.
Defining Quality
In the Preamble to the final rule for 21 CFR Part 111, FDA defines quality for dietary supplements: ‘‘Quality means that the dietary supplement consistently meets the established specifications for identity, purity, strength, and composition and has been manufactured, packaged, labeled, and held under conditions to prevent adulteration under section 402(a)(1), (a)(2), (a)(3), and (a)(4) of the [Federal Food, Drug, and Cosmetic Act].’’
Although warning letters from FDA inspectors repeatedly place emphasis on the five attributes for which specifications must be set (i.e., identity, purity, strength, composition and limits on contaminants), FDA does not provide a specific definition for the meaning of the terms “identity,” “purity,” “strength,” and “composition” in the final rule. According to the Preamble, these terms were not defined because “[t]he exact way in which the dietary supplement industry uses these terms may vary, and defining these terms could limit the flexibility that is needed to accommodate such variations.” Instead of defining the terms, FDA provided interpretations and examples that should be considered when establishing specifications. Table 1 lists the explanatory text from the Preamble (without the specific examples that were provided).
Despite the agency’s intent to allow some flexibility in the use of these terms, the interpretations provided in the Preamble may not necessarily provide the level of clarity desired by the industry. The agency’s interpretations of these terms were set in the context of finished dietary supplements, and may be extended to components, insofar as the specifications of components affect the specifications of the final dietary supplements. USP suggests that further explanation of these terms would be useful, particularly in the context of defining quality attributes for the different classes of components found in dietary supplements. Some other sections of the Preamble to the cGMP final rule clarify the use of these terms as applied to the ingredients (see Table 2).
Terms for Quality Attributes
USP sets standards for the identity, strength, quality, and purity of medicines, food ingredients, and dietary supplements manufactured, distributed and consumed worldwide. USP standards for drugs and dietary supplements are recognized in U.S. federal law and are enforceable by FDA. However, although it is mandatory for drug product manufacturers to comply with USP standards, dietary supplement standards developed by USP are voluntary.
An official USP quality standard for an article (i.e., excipient, dietary ingredient or dietary supplement), referred to as a monograph, sets forth the article’s name, definition, specifications (i.e., tests, test procedures and acceptance criteria) and other requirements related to packaging, storage and labeling. USP monographs include specifications for identity, assay, strength, composition, limits for contaminants, specific tests and/or performance criteria (primarily for finished dosage units). Some of the terms used in USP monographs may differ from the FDA terms, and yet the specifications that they represent are intended to address all of the quality attributes mentioned in FDA’s cGMP requirements, as described in Tables 3 and 4.
In general, USP monographs do not refer to “purity” as a test in a monograph, but rather refer to an “Assay” or “Content” test procedure that can measure the overall purity as referred to in FDA’s cGMPs when applied to an ingredient. The test for strength in a USP monograph is used to measure the amount of a dietary ingredient per unit of measure in a dietary supplement, and it has the same meaning as in FDA’s cGMPs. The term “Composition” in a USP monograph is used to refer to those tests for multiple constituents in a complex dietary ingredient.
USP monographs may include specific tests for an article, such as loss on drying, or residue on ignition. Performance tests such as disintegration or dissolution are performed on a dietary supplement to help ensure that the supplement will release the ingredients properly to make them available for absorption by the body or to exert their function as claimed.
The type of performance test used may be dependent on health considerations (e.g., dissolution test for folic acid in supplements). In conclusion, USP monographs can serve as useful resources to help dietary supplement manufacturers comply with cGMPs when setting quality specifications. Manufacturers can benchmark their tests, analytical procedures, and acceptance criteria against those in USP monographs when setting a specification for a component (e.g., excipient, vitamin, mineral, amino acid, specialty chemical, botanical) or a finished product (e.g., tablet, capsule, or soft gel). USP official standards for dietary supplements offer manufacturers a scientifically valid means of supporting compliance with the specification requirements described in 21 CFR 111.70. Knowledge of FDA’s cGMP requirements related to specifications for dietary supplements—coupled with a robust understanding of how USP monographs support the regulatory regime—can help enhance the quality of the dietary supplement marketplace and protect public health.















